DNA footprinting is a collection of experimental methods
used to describe the sequence specificity of DNA-binding proteins. 3D-footprint
is a database that employs a computational approach in order to analyse
sequence recognition in protein-DNA complexes of known structure, reporting those
molecular contacts that contribute most to recognition. The pipeline followed to
produce each entry in the database can be summarized as a flowchart.
The database contains a selection of 95% non-redundant monomeric complexes updated weekly, plus a
complementary collection of redundant and multimeric complexes. Entries are named by concatenating
the Protein Data Bank (PDB) structure identifier and the protein chains that take part in the complex. For instance,
1cgp_AB, is a dimeric
complex made by protein chains A and B of PDB entry 1cgp.
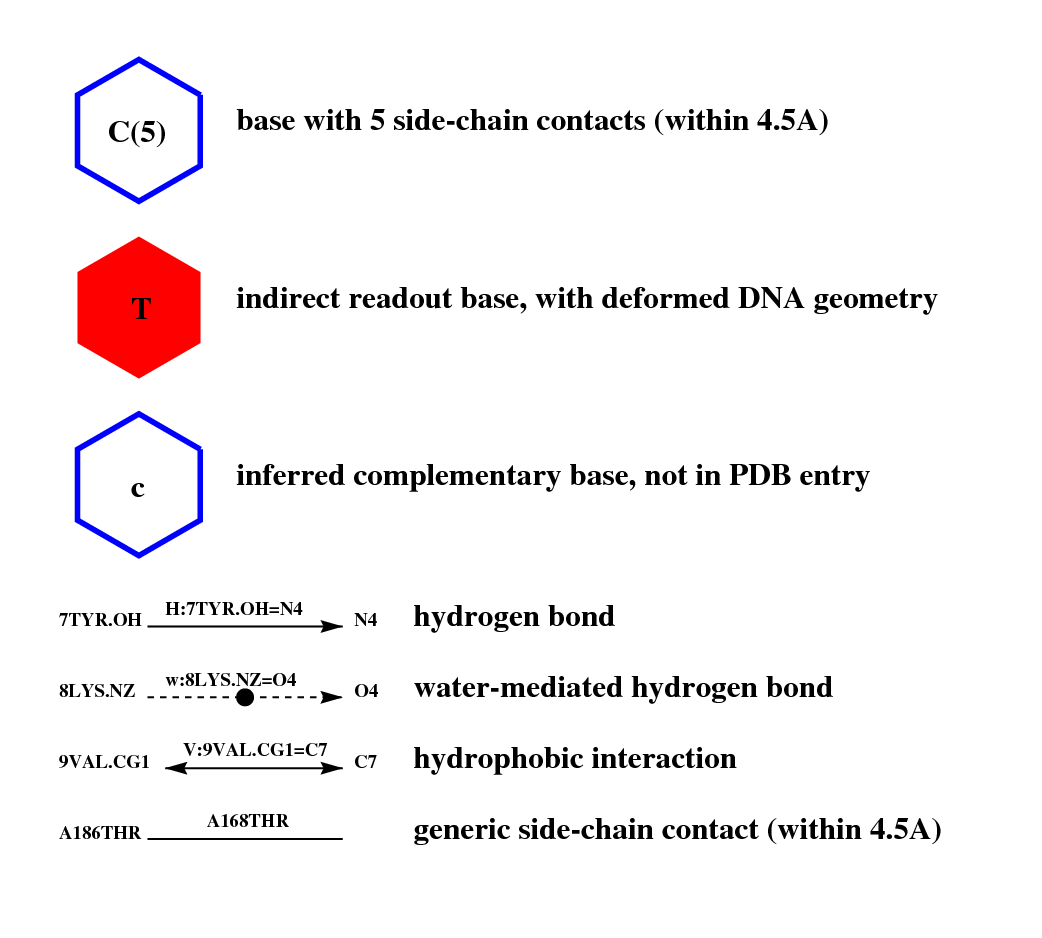
The report for each entry might include:
an interface graph which highlights contacts and nucleotides at the
original interface, captured in a PDB file, responsible for specific DNA discrimination:
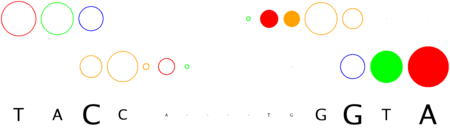
a footprint logo diagram with nitrogen bases depicted as circles of diameter proportional to the
number of side-chain contacts observed at the original interface, with both DNA strands plotted separately. Filled circles
represent indirectly readout bases.
Contact counts can be converted to information content of the resulting contact position weight matrix (see below)
in order to calculate the conservation (height) of the bound DNA sequence:
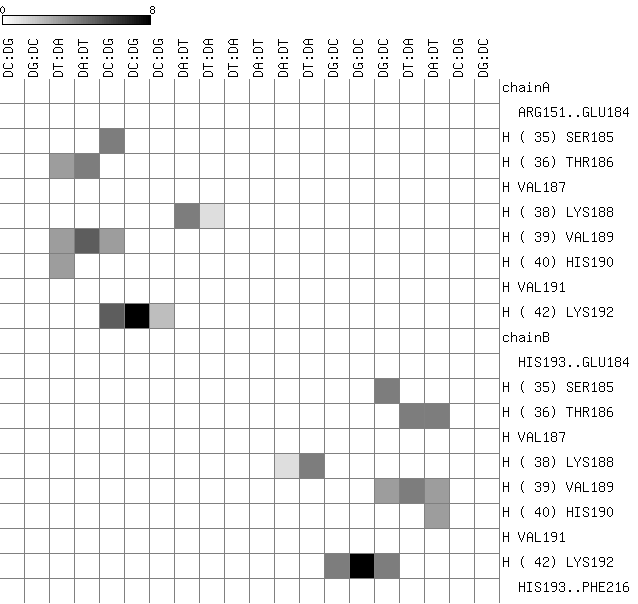
an interface matrix which summarizes the observed contacts between side-chain atoms (vertical axis) and
nitrogen bases of nucleotides (horizontal axis). Most contacted base pairs are dark-coloured, while non-contacted nucleotides are shown in white, according to the scale bar. The secondary structure state of each interface residue is shown in a one-letter code ('H'=helix,'E'=strand,'T'=turn,'C'=coil) next to their primary sequence number, which is parenthesized:
the structure name, a link to the original entry in the PDB, with the name of the protein in parenthesis.
links to multimeric complexes in which a given entry is involved.
Multimeric complexes are usually more relevant in biological terms and have more selective binding motifs.
links to a list of redundant complexes, with % of protein identity > 95,
which are considered to be represented by the entry in question, or otherwise a link to a non-redundant reference complex.
Redundant complexes can be important as often they provide more accurate specificity estimates, and for this reason the redundant entry
with the highest information content is indicated by an arrow .
a reference complex that represents a cluster of redundant entries
that are at least 95% identical in protein sequence.
the protein sequence of the complex in question with the list of (upper-case) interface residues plotted in the
interface graph:
Interface residues are numbered as they appear in the protein sequence. Original PDB residue numbers, which are preferred in interface graphs,
can be mapped using the list of :
the interface signature is a string of concatenated interface residues:
RKIQNM
estimated binding specificities as position weight matrices (PWMs) of two types:
contact and readout. Contact PWMs are calculated by adding contacts between side-chains and nitrogen bases,
assuming that the DNA molecule in the complex is the cognate sequence, after Morozov's approach.
Readout PWMs are instead derived both from i) the array of scored atomic interactions at the interface (direct readout),
and ii) the set of sequence-dependent deformations inferred from the DNA coordinates (indirect readout), that are blended
by applying the DNAPROT algorithm. In general, both approaches provide similar PWMs
(see the DNAPROT paper), and mean PWMs and sequence logos are provided for each entry.
However, readout PWMs are evaluated as unreliable when the number of interface atomic contacts is less than 5 or when the
cognate DNA sequence has an associated PWM score below the top 80% (see benchmark)
and in those cases (marked with ) the provided specificity estimates correspond
exclusively to contact PWMs. In either case PWMs are evaluated in terms of their information content (IC),
which can be read as a measurement of binding specificity, and those PWMs with less than 4 informative columns are said to be
unspecific and discarded.
These PWMs can be used to scan DNA sequences or even genomes following the scan! link, that takes you to a RSA-Tools form where you can paste or upload the sequences to be scanned.
a table with related DNA sequences reported in the literature,
available only for non-redundant entries, such as binding sites or consensus sequences found in abstracts of relevant reserch papers.
These data can be helpful to evaluate the binding preferences of related proteins or the range of variant sites bound by the same
transcription factor. Sites are reported with associated STAMP E-values that measure
local similarity to related multimeric and redundant complexes. Please note that it might be necessary to take complementary sequences before comparing sites:
a dendrogram of similar interfaces, available only for non-redundant entries.
This distance tree is based on the observed structural similarity between DNA-binding domains, and interfaces are aligned showing
pairs of interacting amino acids chains and nucleotide bases. Only N-ring (purine/pyrimidine) heavy atom contacts less than 4.5Å
away are considered here. For instance, RA stands for arginine in
contact with adenine (only bases are colored).
Note that this alignment format allows only one base (the closest one) per interface residue, overlooking the cases in which a single
residue contacts several bases.
SCOP superfamilies of reported complexes can be seen by mousing over the tree leaves,
while structure-based sequence logos are clickable L links:
RCNTFTMA--RAHGNT--SA------YA-- L +3f27_D
+-1
KCNT--MAMA--EGSAACNT---------- L +-3 +2lef_A
! !
RCNTFTMA--RA--NGSCST------YA-- L +-4 +1gt0_D
! !
RTNGFTIA--RA--NASGST------YAPGL +-5 +-1j47_A
! !
RTSAYCMT------FT-------------- L +-----6 +---1j5n_A
! !
----YC--VT----FC--SA---------- L ! ! +1ckt_A
--7 +---2
RC--YTMALTRA--VCTGAA--VCEC---- L ! +1qrv_A
!
RCNAFTIA--RG--NC--SALT----YAKTL +----------2gzk_A
The same data can be displayed as a matrix of homologous interface contacts, in which
MAMMOTH structural alignment -ln(Evalues) are shown.
This is a vertical alignment with one column per complex, where each column shows the aligned equivalent protein residue and the
contacted nucleotide. Residues marked with * are interface residues in the reference complex. For instance:
0208 E* ECRG------SCKG--HG----RG--EC 0.91
means that residue E(Glu) 208 is aligned to 7 equivalent residues, two of which are E that contact C nucleotides.
 .
.
 ) the provided specificity estimates correspond
exclusively to contact PWMs. In either case PWMs are evaluated in terms of their information content (IC),
which can be read as a measurement of binding specificity, and those PWMs with less than 4 informative columns are said to be
unspecific and discarded.
) the provided specificity estimates correspond
exclusively to contact PWMs. In either case PWMs are evaluated in terms of their information content (IC),
which can be read as a measurement of binding specificity, and those PWMs with less than 4 informative columns are said to be
unspecific and discarded.
 position weight matrix (PWM)")